Background and Overview
What is live-cell imaging?
Live-cell imaging, as the name suggests, is a technique, whereby the cells are imaged when they are alive, typically in the growth medium. It is a powerful imaging approach that provides spatiotemporal changes of subcellular events in real-time, thus providing deeper insights into cellular structure and functions. In recent years, live-cell imaging has become a requisite analytical tool for addressing important questions in several areas of biological and biomedical sciences including cancer research, neuroscience, cell biology, developmental biology, and pharmacology1-2.
What are the advantages of studying living cells over fixed cells?
In cellular imaging, two major paradigms exist: i)static imaging with “fixed cells” and ii)dynamic imaging with “live cells”. While “fixed cells” are representative of static cells that remain unchanged over time, “live cells” represent dynamic cells that move and change over time3. Traditional static imaging tools involve “fixation” of cells and tissues so that cellular/tissue components are preserved in a “life-like state”. Although cells die during physical and chemical fixation, their shape, contents, broad patterns, networks, and proteins are mostly conserved for imaging purposes. To detect intracellular antigens, cells can be also permeabilized post-fixation, i.e. cellular membrane lipids can be effectively removed to provide access to large molecules such as antibodies. Compared to live cells, fixed cells are easier to prepare and stain. They can be preserved in the original condition for months or years by simply mounting on a cover slip, allowing greater ease for imaging and detailed analysis. Despite these advantages, imaging data obtained using fixed cells are not representative of actual cell behavior and characteristics of a living system. To obtain more biologically relevant data, imaging over time, also known as time-lapse imaging, is essential. Instead of providing just the “snapshot” of a cell’s current state, live-cell imaging transforms snapshots to movies, allowing for the visualization and quantification of dynamic cellular processes in real-time and over time. With a growing surge of interest in the investigation of dynamic processes such as migration, cell development, and intracellular trafficking, live-cell imaging has become an indispensable tool for understanding the dynamic molecular events in single cells and cellular networks both in vitro and in vivo.
Benefits of live-cell imaging
Live-cell imaging offers the following advantages4:
- Real-time monitoring of living cells enables visualization of transient events that may be missed in end-point assays and thus, reveals optimal time points for end-point assays
- Live-cell imaging allows studying cellular structures in their native environment making them less prone to experimental artifacts as compared to fixed cell microscopy
- It allows simultaneous, real-time tracking of the localization and transport of cellular biomolecules along with the progression of multiple pathways
- Lateral, axial, and temporal image acquisition enables the creation of 4D images and data
History and Methods of Live-Cell Imaging
Ever since the invention of microscope by spectacle-makers father and son duo, Hans and Zacharias Janssen, the scientists have been exposed to the fascinating world of microorganisms. Initially, the researchers used to record their findings using simple hand-drawn images. However, further urge for documentation fostered the development of microcinematography, a technique used to create short films showing the movement of bacteria or other microbes. Today, visualization technology in life sciences has progressed to such an extent that using live-cell imaging, we can record detailed videos of cells and study the subcellular structures in real-time. In this section, we will briefly describe the various phases of development in the field of cellular imaging (Table 1) starting from microcinematography to various cutting-edge technologies used in modern live-cell imaging2,5.
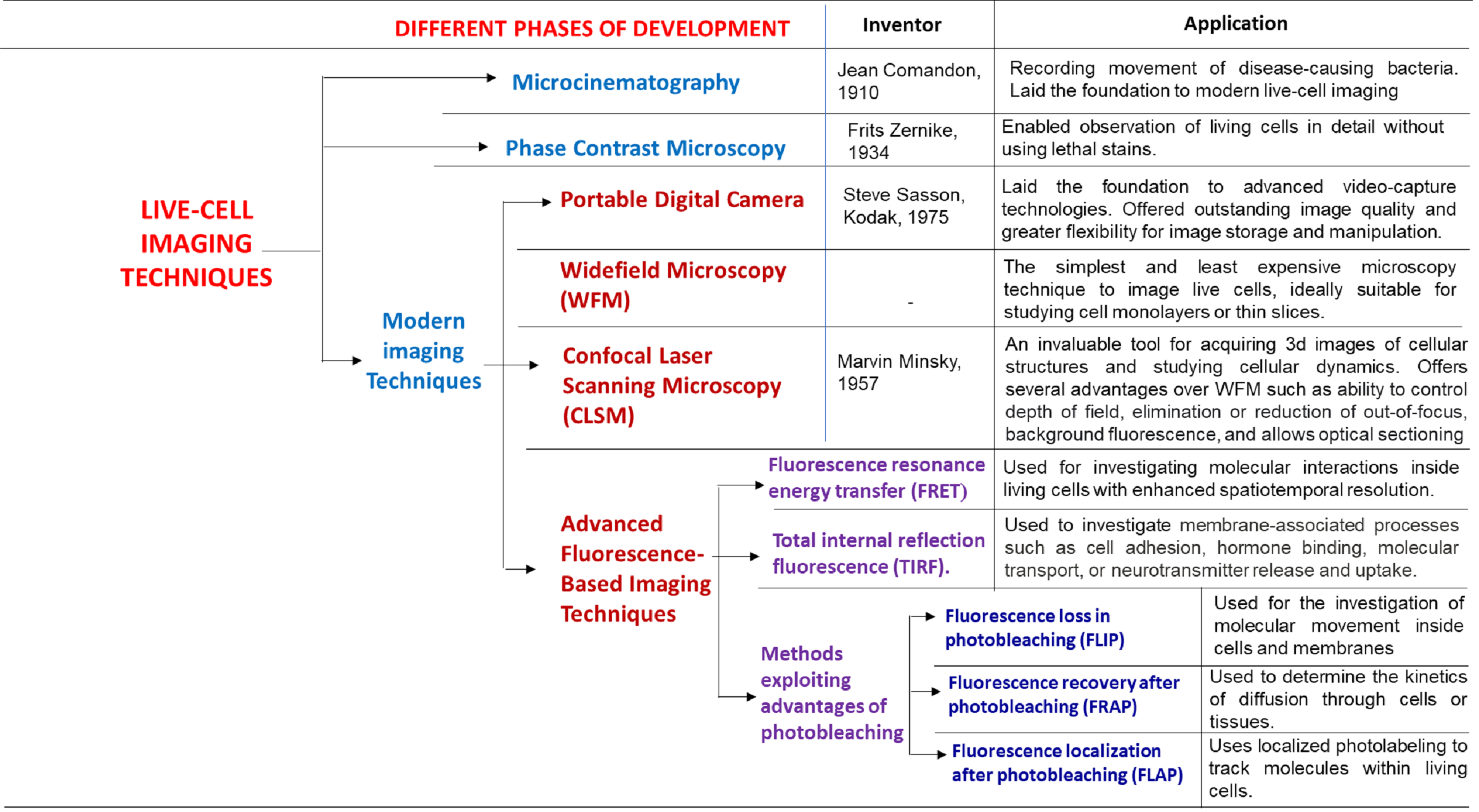
Microcinematography
In the early nineteenth century, researchers started to get ideas from the emerging film and cinema industry on how live cells could be recorded2. In 1910, the French biologist and filmmaker, Jean Comandon made his pioneering contribution to the field of microcinematography. He used a giant cinema camera affixed to an ultramicroscope to record the movement of Treponema pallidum, a spirochaete bacterium with various subspecies that causes diseases such as syphilis, bezel, and yaws6. He showed that the movement of disease-causing bacteria is unique and different from the non-disease-causing ones. He strongly believed that recording cellular movement on film could improve the physicians’ understandings of certain diseases, facilitating more accurate diagnosis. He continued to produce many other biological and medical films of cells, some of which were even shown in Paris theaters in between the newsreels and entertainment news2,7. These movies played a pivotal role in educating common people about cells and molecules, stimulating their interest and excitement about the then-unknown microbial world. Subsequently, scientists came up with more advanced video-capture technologies such as time-lapse microcinematography, which involved recording images at evenly spaced intervals and playing them at a higher speed thereafter to accelerate the observed movements. Such modification not only transformed the previously undetectable, slow changes to detectable and visible ones but also laid the foundation for today’s live-cell imaging.
Phase contrast microscopy
A brief overview of phase contrast
An enormous leap in dynamic cellular imaging came in 1934 with the invention of phase contrast microscopy, a light microscopy technique used to enhance contrast in a translucent specimen without staining it8-9. This technique employs an optical mechanism to translate the differences in phase of the light transmitted through or reflected by the specimen to corresponding changes in amplitude, which can be visualized as changes in image contrast. Although phase shifts themselves are invisible, they become visible when presented as brightness variations10.
Historical perspective
The first phase contrast microscope was designed by the Dutch physicist, Frits Zernike, with the help of Zeiss Optical Works in Jena, Germany11-12. In 1941, a biologist named Kurt Michel from Carl Zeiss company, used this newly invented phase contrast microscope to record the time-lapse movie of the prophase stage during meiosis I in spermatocytes of Psophus stridulus, commonly known as rattle grasshopper13. He developed a micro-cinematographic system with a 35 mm Debrie camera and started documenting the mitotic process in cell division using time-lapse cinematography. In 1945, realizing the importance of this technique, the majority of the microscope manufacturers rushed to produce their own microscopes with the enhanced specimen illumination mode, designed by Zernike. In 1958, Michel’s film entitled, “Meiosis in the spermatogenesis of Posphus stridulus L.’ was published by the Institute of Scientific Cinematography in Gottingen. This educational documentary had been used in academic teaching for many years, thereafter. The invention of the phase contrast microscope had been such a remarkable advancement in biological research that Frits Zernike was awarded the 1953 Nobel Prize in Physics. To further improve cellular imaging, more advanced phase contrast techniques such as Hoffman modulation contrast (HMC) microscopy and differential interference contrast (DIC) microscopy were developed. Modern phase contrast objectives are equipped with several contrast-enhancing techniques that can effectively alter absorption levels of the surrounding illumination, generating a broad spectrum of specimen contrast and background intensity.
Quantitative phase contrast microscopy
This technique relies on the quantification of phase shift that occurs when light waves pass through a more optically dense object14-16. Currently, live-cell imaging is largely restricted to observing cells on a single plane due to the narrow focal depths of conventional microscopy. This limitation can be overcome by using quantitative phase contrast microscopy, which allows creating and focusing images on different focal planes from a single exposure. Integration of rotational scanning feature further allows acquiring 3D time-lapse images at high resolution17-18. This technique is primarily used for non-invasive live-cell imaging and automated cell culture analysis.
Advantages of phase contrast microscopy
Before the introduction of phase contrast microscopy, it was difficult to observe living cells because the latter, being translucent, required staining to be visible under a traditional light microscope. Unfortunately, fixation and staining would kill the cells. For the first time, phase contrast microscopy made it possible to observe living cells in detail without using lethal stains and study their proliferation through cell division.
Limitations
Phase contrast microscopy lacks the ability to detect specific proteins or other organic compounds constituting the cellular backbone. This shortcoming necessitated the development of synthetic and organic fluorophores (fluorescent dyes or stains) that could efficiently label the subcellular structures as well as various compounds (antibodies, proteins, or small molecules) present in a cell, allowing their visualization by fluorescence microscopy.
Digital camera and modern imaging techniques
The next revolutionary milestone in live-cell imaging was the invention of a user-friendly, self-contained (portable) digital camera by Steve Sasson at Kodak in 19752, 19. This newly discovered camera offered outstanding image quality and greater flexibility for image storage and manipulation. The advent of digital cameras fostered the development of several microscopic techniques such as widefield microscopy (WFM), confocal laser scanning microscopy (CLSM), and a variety of new fluorescence-based imaging techniques such as FRET, TIRF, FRAP, and FLIP. These cutting-edge techniques replaced the traditional biophysical, biochemical, and in vitro screening methods such as chromatography, co-immunoprecipitation, and phage display, while making it possible for the scientists to analyze the movement and interactions of the protein partners inside individual living cells.
Widefield microscopy (WFM)
It is one of the simplest and least expensive microscopy techniques to image live cells, ideally suitable for studying cell monolayers or thin slices1-2, 20.
Principle: in this technique, the entire specimen of interest is illuminated. Generally, mercury arc lamps and xenon lamps are used as the source of light, though light-emitting diodes (LED) have started being used in recent years. The resulting image is either viewed by the observer or a camera, which can be also connected to a computer monitor to allow external video display.
Advantages:
- This technique is simple to use and much less expensive than a confocal microscope
- Confocal quality images of thin samples can be obtained by applying deconvolution and other post-processing techniques
- It can be used for imaging biological samples that are small, blurry, and/or alive
- This technique works well for dynamic situations where moving specimens are imaged
- It offers easy housing and maintenance and functions very well for thin samples
Limitations:
- This technique is not suitable for imaging thick specimens
Confocal laser scanning microscopy (CLSM)
This optical imaging technique enhances the optical resolution and contrast of a micrograph using a spatial pinhole to reject out-of-focus light from the detector so that it does not cause blur to the images being recorded21-22.
Principle: in CLSM, both illumination and detection optics are focused on the same diffraction-limited spot in the test sample, the only spot imaged by a detector during a confocal scan. To produce a complete image of an object, this spot is moved over the sample and the emitted photons are captured pixel-by-pixel23.
Historical perspective: in 1957, Marvin Minsky, a postdoctoral researcher at Harvard University, patented the basic principle of confocal microscopy24-26. With a goal to image dynamic processes in living systems, Minsky sought to image the neural networks in an unstained sample of brain tissues. However, his invention did not bear fruit at that time due to the lack of i) intense light sources; and ii) computer horsepower necessary to process such large amounts of data. In 1960, M. David Egger and Mojmir Petran took Minsky’s work a step forward by fabricating a multiple-beam confocal microscope, which utilized a spinning disk known as Nipkow for investigating brain sections and ganglion cells. Eventually, Egger extended this technique to develop the first mechanically scanned confocal microscope, which was then used to publish the first identifiable images of cells in 1973. As later scientific advancements paved the way to the cutting-edge computer and laser technologies as well as digital manipulation of images using algorithms, the field of confocal microscopy witnessed phenomenal growth. In the late 1970s and 1980s, practically usable confocal microscopes were conceptualized and developed by a series of scientists including G. Fred Brakenhoff (1979), Collin Sheppard, Tony Wilson, Brad Amos, and John White (1980s). The first commercial confocal microscope appeared in 1987. Soon after, in the 1990s, advancements in the field of optics and electronics facilitated the development of more stable and powerful lasers, high-throughput fiber optics, scanning mirror units with higher efficiency, improved thin-film dielectric coatings, and low-noise detectors25. Concomitantly, advances in organic chemistry made it possible to synthesize a variety of fluorochromes carefully matched to the laser excitation lines. In the late 1990s, further improvement in computer processing speeds, display enhancement, and the emergence of large-volume storage technologies, laid the foundation for the modern confocal microscope. There are four types of confocal microscopes that are commercially available: i) confocal laser scanning microscopes (CLSM); ii) spinning disc or Nipkow disc confocal microscope; iii) dual-spinning disc confocal microscope; and iv) programmable array microscope27-28. Today, confocal microscopy has become an indispensable tool for producing 3D images of cellular structures and investigation of cellular dynamics.
Advantages: confocal microscopy offers several advantages over conventional widefield microscopy, such as:
- It permits selection of user-defined regions of interest during the scan, eliminating the necessity to scan the entire specimen
- It allows optical sectioning. A confocal microscope has the unique capability to collect serial optical sections from thick specimens, which facilitates the three-dimensional reconstruction of a sample from high-resolution stacks of images
- It can control the depth of the field. In a conventional microscope, light travels through the sample as far as it can penetrate the specimen. However, a confocal microscope can attain a controlled, and highly limited depth of field by focusing only a smaller beam of light at one narrow depth level at a time
- It can also eliminate or reduce much of the background fluorescence that is out-of-focusand may lead to image degradation
Limitations:
- Confocal microscopes are very expensive to manufacture and purchase
- They provide a relatively smaller field of view
- They have only a limited number of excitation wavelengths, with very narrow bands
Advanced fluorescence-based imaging techniques
In recent years, a variety of advanced fluorescence-based imaging techniques are used in live-cell imaging. An overview of these techniques is summarized as follows.
Fluorescence resonance energy transfer (FRET). It is a distance-dependent physical process that involves the non-radiative transfer of energy from an excited molecular fluorophore (the donor) to another fluorophore (the acceptor) via intermolecular long-range dipole-dipole coupling29. The most widely used donor and acceptor fluorophores for FRET experiments come from a class of autofluorescent proteins, such as the green fluorescent proteins or GFPs. Recent advances in fluorescence microscopy, combined with the development of new fluorescent probes have made FRET an important tool for investigating molecular interactions inside living cells with enhanced spatiotemporal resolution.
Total internal reflection fluorescence (TIRF). TIRF is a special technique in fluorescence microscopy, invented in the early 1980s by Daniel Axelrod at the University of Michigan30-32. It exploits the unique properties of an induced, evanescent wave or field in a small region of the specimen that is immediately adjacent to the interface between two media having different refractive indices (e.g. glass/specimen interface). This modality can be used to selectively excite fluorophores bound to the cell surface, while non-bound molecules are not excited and do not fluoresce. It offers several benefits over widefield and confocal fluorescence microscopy:
- This method allows clear visualization of structures due to its reduced background noise, and subsequently, enhanced signal to noise ratio (SNR)
- It decreases the blurring effects due to rejection of out-of-focus fluorescence
- Exposure of cells to substantially smaller amounts of lights results in lower phototoxicity
TIRF microscopy is a promising modality for the investigation of membrane-associated processes, such as cell adhesion, hormone binding, molecular transport phenomenon, and various exocytotic and endocytotic processes such as neurotransmitter release and uptake.
Methods exploiting advantages of photobleaching. When a fluorophore is excited with an intense laser source or exposed to the source for a prolonged duration, the fluorescent molecules lose their ability to emit light. This process is known as photobleaching. Bleached fluorophores permanently lose their capability to fluoresce and act as imaging markers. Interestingly, certain techniques such as FLIP, FRAP, and FLAP take advantage of this bleaching33-34.
Fluorescence loss in photobleaching (FLIP). This technique is used to investigate molecular movement inside cells and membranes35-36. In this modality, the cell membrane is labeled with some fluorescent dye (e.g. GFP) to enable visualization. Thereafter, a specific area of this labeled section is illuminated repeatedly with a high-power laser beam (e.g. a beam of CLSM) to induce photobleaching. Images are captured with reduced laser power and time lag between the bleaches. The amount of fluorescence from the selected region is then measured over time to get information on protein dynamics in various cellular regions.
Fluorescence recovery after photobleaching (FRAP). This technique is used to determine the kinetics of diffusion through cells or tissues. In this method, a specific area of a cell or tissue is photobleached by high-power laser light, removing fluorescence from this area. Because this technique requires the fluorescent molecules to move around freely, the selected region of interest is typically a cell membrane or an area within the cell where diffusion occurs (e.g. nucleus). Fluorescence in the bleached area gradually recuperates as bleached fluorophores move out, while intact, unbleached fluorophores move in37. This technique has promising implications in the studies of cell membrane diffusion, and protein binding, as well as characterization of hydrophobicity or hydrophilicity of a surface.
Fluorescence recovery after photobleaching (FLAP). FLAP is a relatively new method, which employs localized photolabeling to track molecules within living cells33. In this method, the biomolecule of interest is tagged with two fluorescent markers: i) a control fluorophore, which remains intact serving as a reference; and ii) a target fluorophore that is rapidly photobleached at the chosen location. These fluorochromes can be measured either independently or simultaneously. By simple image differentiation, it is possible to locate the distribution of cellular molecules and characterize their mobility.
Conclusion and Future Perspectives
The continued development of new microscopy techniques, optical biosensors, and sophisticated image analysis pipelines have paved the way to numerous live-cell imaging technologies that provide in-depth temporal profiling of various cellular processes, down to the molecular level. In recent years, live-cell imaging has become an invaluable tool in preclinical drug discovery38. Real-time, image-based evaluation of drug response upon target activity and pathophysiology in vitro as well as in vivo may accelerate the timeline for drug development, decrease cost, and provide novel insights into the molecular mechanisms of drug action. In the current scientific panorama, where the outbreak of the COVID-19 pandemic caused by SARS-CoV-2 coronavirus has severely affected the global community, live-cell imaging has emerged as a highly appealing tool for studying the receptor binding, cellular uptake, and intracellular trafficking of SARS-CoV-2 in fixed and live samples with high specificity39. More recently, the launch of remote live imaging systems has allowed COVID-19 researchers to observe and analyze their cells without entering the lab while saving time and lowering the risks associated with performing COVID-19 research40. While the advantages and necessity of live-cell imaging are well-appreciated, a key challenge underlying this technique is keeping the cells healthy and alive throughout the observation period. To obtain the best image quality while maintaining healthy cells, a myriad of factors needs to be considered. These include imaging modality, media, temperature, pH, humidity, osmolality, wavelength of light, and total photon dose used during experimentation. We anticipate that with incessant technological advancements and the design of new fluorescent probes, the challenges and limitations associated with live-cell imaging will dwindle over time.
----------
All CytoSMART devices are incubator-friendly, allowing to study cells in their favorable culture environment. Learn more about the CytoSMART imaging solutions here.
----------
Related Products
There are currently no products tagged to this resource.