Background and Overview
Chimeric antigen receptor (CAR) T cell therapy is a rapidly emerging treatment modality that uses genetically engineered T cells to specifically target and kill cancer cells1-4. As its name suggests, the backbone of CAR T cell therapy is T cells, a type of leukocyte (white blood cell), that plays a key role in the adaptive immune response. The invention of CAR T cell-based immunotherapy has revolutionized the field of personalized cancer treatment. CAR T cells can be produced from either T cells of a patient’s own blood (autologous) or that of another healthy donor (allogenic)5. However, for most currently available therapies autologous cells are used. Once isolated from a patient, these T cells are genetically modified in a laboratory to express CARs that are specific to a particular tumor antigen. The CARs are designed to endow these T cells with highly specific homing abilities so that after reinfusion into the patient, they can act as a “living drug” against cancer cells. The engineered receptors (CAR) allow these T cells to easily recognize their targeted antigen on a tumor cell, bind to it, and exert selective cytotoxicity (Figure 1). In Greek mythology, “Chimera” also called Chimaera was the name given to a fire-breathing, female monster that resembled a lion in the front, a goat in the middle, and a dragon behind6. In biology, the term “chimera” is used to depict a single organism that is made up of genetically different cells or a hybrid protein synthesized by splicing different pieces of a genetic code together7. The term “chimeric” has been attributed to these engineered receptors because they combine both antigen-binding and T cell-activating functions into a single receptor.
Cancer Immunotherapy Using CAR T Cells
Innate immunity vs adaptive immunity
The immune system of our body represents a large, complex network of cells, tissues, organs, and substances that work together to protect us against microbial infection and diseases8. It is comprised of two main parts: i) the innate (natural) immune system and ii) the adaptive (specialized) immune system. Innate immunity or natural immunity is the body’s first line of defense, which triggers a fast yet non-specific, inflammatory response immediately or within hours of an antigen’s appearance in the body. It encompasses physical barriers of our body such as the skin, chemicals in the blood, and immune cells that identify foreign microorganisms as 'non-self' and trigger a response to destroy these disease-causing agents. The adaptive immune system is activated when the innate immune system fails to control infection. Unlike innate immunity, adaptive or acquired immunity entails an antigen-specific immune response that is highly adaptive to a specific pathogen. Thus, adaptive immune responses are slower and more complicated, yet more accurate than innate immune responses. Adaptive immunity also creates immunological memory after an initial response to a specific pathogen so that when it encounters the same pathogen in the future, it responds faster and more efficiently. The adaptive immune system involves i) B lymphocytes or B cells and ii) T lymphocytes or T cells9. B cells, which originate and mature in the bone marrow, act as the key players in antibody-mediated immunity. T cells, which originate in the bone marrow and mature in the thymus, have several important functions. These include stimulating the B cells to generate antibodies to fight infection, directly killing infected host cells, and producing cytokines in response to an immune challenge.
Cancer immunotherapy using CAR T cells
Engineering immune cells to fight cancer
The immune system not only defends our body against foreign intruders (e.g. disease-causing microorganisms) but also plays a critical role in protecting our body from malignancies10-12. Cells start to grow uncontrollably and become cancerous as mutations accumulate in the various genes regulating cell proliferation. DNA damage is an important hallmark of cancer development and progression. Directed by the damaged DNA in cancer cells, the mutated cells frequently produce abnormal proteins known as tumor antigens. Subsequently, our immune system marks cancer cells as ‘non-self’ and triggers antitumor immune responses to inhibit the growth and development of malignant tumors. Interestingly, cancer cells seem to possess certain mechanisms that allow them to bypass the normal immune system response. With CAR T cell therapy, scientists can add engineered receptors onto the surface of T cells, called the chimeric antigen receptor or CAR, which enables them to identify and selectively destroy cancer cells.
The structure of CAR T cells
CARs are modular, with a synthetic receptor consisting of three main regions5, 13-16 :
- An extracellular ligand-binding domain derived from the variable regions of a monoclonal antibody combined as a single-chain fragment variable (scFv)17. This region is responsible for recognizing a specific target antigen present on the surface of cancer cells.
- A transmembrane domain and a hinge/extracellular spacer are responsible for linking the other two regions of a CAR while augmenting the overall flexibility, stability, and dimerization of the construct.
- An intracellular T cell signaling domain is responsible for the transmission of a signal after an antigen binds to the external ligand-binding domain. Most of the CAR construct modifications are performed in this region to further improve the signal transmission capacity of a specific CAR.
CAR T cells can be both CD4+ and CD8+ though the efficacy of any adoptive cell therapy is mostly attributed to CD8 T cells. While CD4 cells assist other blood cells to produce an immune response, the CD8 cells are a group of cytotoxic T cells that causes cell death via lysis or apoptosis. According to recent reports, a 1:1 ratio of CD4:CD8 CAR T cells confers enhanced antitumor reactivity in vivo, implying synergistic antitumor efficacy of the two subsets18-19.
How CAR T cells work
CAR T cells work by circulating in a patient’s body and looking for cells that possess the antigen programmed into the CAR protein, for example, cancer cells. As CAR T cells encounter their target antigen on a cell, they bind to it and become activated. The activated CAR T cell then proceeds to proliferate, release cytokines, and exert selective toxicity on cancer cells.
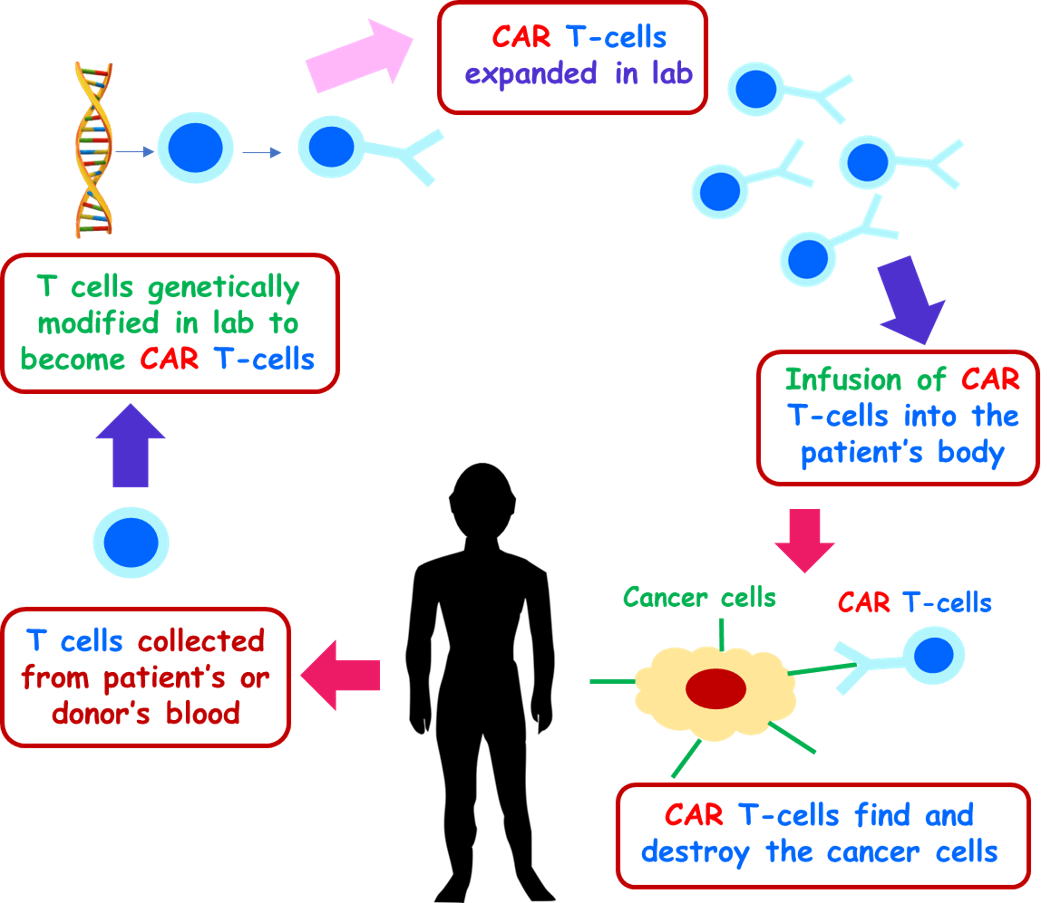
CAR T cell therapy: procedure details
Patient selection
To be eligible for CAR T cell therapy, a patient must fulfill certain criteria20. For instance, the tumor under investigation must express a CAR-specific target, such as BCMA, CD19, CD22, CD23, ROR1, etc. In addition, the patient must have an ample number of T cells for collection, adequate organ function, and status, and should not be suffering from active, uncontrolled infections (e.g. hepatitis B, hepatitis C or HIV) or clinically relevant comorbidities (e.g. selected cardiovascular, neurological or immune disorders).
Collecting cells from a patient
In this therapeutic process, the T cells are collected from a patient through apheresis, a process, which involves circulating the patient’s blood through a machine that can filter out T cells and send the remaining blood back to the person12.
Manufacturing CAR T cells
Post-isolation, the T cells are sent to a manufacturer for producing CAR T cells. To make CAR T cells, the patient’s normal T cells are activated, multiplied, and infected with a virus, which results in a genetic alteration that adds the CAR to the patient’s T cells. These cells are then grown and expanded in vitro for clinical application. It may take several weeks to accrue enough CAR T cells needed for the intended therapeutic usage.
Infusing CAR T cells back to the patient
Before CAR T cell infusion, the patient is given a short course of chemotherapy over 2-3 days using a technique called lymphodepletion so that the patient’s immune system does not consider infused CAR T cells as “abnormal” and eliminates them. The CAR T cells are then returned back to the patient through intravenous infusion.
Recovering phase
The risk/recovery period after CAR T cell therapy is usually around 2-3 months. As CAR T cells multiply within the patient’s body, they can cause serious side effects, including cytokine release syndromes and immune effector cell-associated neurotoxicity21. As per the guidelines of the U.S. Food and Drug Administration (FDA), the patients treated with CAR T cell therapy need to be closely monitored during the initial 20-day acute recovery period.
Which types of cancer are treated by CAR T cell therapy?
CAR T cell therapy is primarily used for the treatment of various hematological cancers such as diffuse large B cell lymphoma (DLBCL), mantle cell lymphoma, follicular lymphoma, multiple myeloma, and B-cell acute lymphoblastic leukemia (ALL) in pediatric as well as young adult patients up to the age of 2512. Additionally, several clinical trials and research studies have evaluated the efficacy of CAR T cell therapies against solid tumors including ovarian22-23, breast24-25, prostate26, renal27, gastric28, lung29, liver30, and colorectal cancer31. The effectiveness of CAR T cell therapy in solid tumors is somewhat limited as compared to hematological tumors because when returned to the patient, CAR T cells are exposed to the bloodstream and lymphatic system. Subsequently, they have greater contact with the blood tumor cells, whereas in solid tumors, CAR T cells may not be able to infiltrate tumor tissue through the vascular endothelium. The heterogeneity of tumor antigens and immunosuppressive tumor milieu also undermines the efficacy of CAR T cell therapy against solid tumors32.
The Evolution of CAR T Cell Therapy: a Brief History
The first chimeric receptors, which comprised parts of an antibody and the T cell receptor, were depicted in 1987 by Yoshihisa Kuwana and colleagues at the Institute of Comprehensive Medical Science in Aichi, Japan33. The first CAR T cells were developed by Israeli immunologists, Giden Gross and Zelig Eshhar, in between 1989-199334. Initially, called “T-bodies”, these early approaches entailed combining an antibody’s ability to specifically bind to different targets with the constant domains of T cell receptors alpha and beta (TCR-α and TCR-β)35. In 1991, Arthur Weiss at the University of California, San Francisco demonstrated that chimeric receptors containing the intracellular signaling domain of CD3ζ can activate T cell signaling36. These results prompted CD3ζ intracellular domains to be added to the first-generation chimeric receptors with antibody-like extracellular domains, most commonly an scFV domain, as well as proteins such as CD4. In the mid-1990s, a first-generation CAR containing a CD4 extracellular domain and a CD3ζ intracellular domain was used in the first clinical trial of CAR T cells by the biotechnology company, Cell Genesys37. In this trial, adoptively transferred T cells were allowed to target HIV-infected cells. Unfortunately, these first-generation CARs failed to exhibit any clinical improvement. Similarly, some early clinical trials of CAR T cells in solid tumors also failed because the transferred T cells did not show long-term persistence in the patient’s body. In the early 2000s, Dr. Michel Sadelain and colleagues demonstrated that if co-stimulatory domains such as CD28 or 4-1BB (CD137) were added to the first-generation CAR’s CD3ζ intracellular domain, these constructs showed longer persistence and improved tumor clearance in preclinical models, setting the foundation stone for second-generation CARs38. In the early 2010s, clinical trials involving the second generation CD 19-targeted CAR T cells were initiated independently by the investigators at the National Cancer Institute (NCI), University of Pennsylvania (UPENN), and Memorial Sloan Kettering Cancer Center37. CD19 is a B-lineage-specific transmembrane glycoprotein overexpressed on essentially all B-lineage leukemias and lymphomas, including the pre-B acute lymphoblastic leukemias (pre-B ALLs), chronic lymphocytic leukemia (CLL), and lymphomasnormal B cells39. These CD19-targeted CAR T cells resulted in complete to partial remissions in many patients and thus, established the clinical effectiveness of CAR T cell therapy. The success of these trials paved the way to the FDA approval of the first CAR T cell-based product, Tisagenleclucel (Kymriah) in 2017. The important clinical trials and publications that led to tisagenlecleucel approval are presented in Table 1.
Table 1 | Tisagenleclucel (Kymriah): pivotal clinical trials leading to FDA approval.
| Underlying malignancy | Primary study population | Sponsored by | Study location | Phase of clinical trial | No. of patients infused | Result: CR/CRi at 3 months* (%) | Reference(s) |
| Relapsed/ Refractory Chronic Lymphocytic Leukemia (CLL) | Adult | UPENN | Abramson Cancer Center (ACC)/UPENN | Pilot/I | 14 | 29 | 40-42 |
| Relapsed/ Refractory B-cell acute lymphoblastic leukemia (B-ALL) | Children | UPENN | Children’s Hospital of Philadelphia (CHOP)/UPENN | I/IIa** | 30 | 90 | 43 |
| Children | Novartis | United States | II | 29 | 69 | 44 | |
| Children | Novartis | Multinational | II | 75 | 81 | 45 | |
| Relapsed/ Refractory Diffuse Large B Cell lymphoma (DLBCL) | Adult | UPENN | ACC/UPENN | IIa | 14 | 43* | 46 |
| Adult | Novartis | Multinational | II | 93 | 40 | 47 |
* CR and CRi denote complete remission and remission with incomplete hematologic recovery, respectively.
** Phase IIa clinical trials are proof-of-concept studies conducted on a relatively small number of patients. Such studies are used to evaluate the effectiveness of a particular product against a placebo or other positive controls for a particular clinical indication.
Clinical Applications of CAR T Cell Therapy
Approved CAR T cell therapies by the US-FDA
To date, five CAR T cell-based products have been approved by the US-FDA for the treatment of different types of hematological malignancies5, listed in Table 2. These include: Tisagenlecleucel (Kymriah), Axicabtagene ciloleucel (Yescarta), Brexucabtagene autoleucel (Tecartus), Lisocabtagene maraleucel (Breyanzi), and Idecabtagene vicleucel (Abecma). Among these, the European Commission (EC) has granted marketing authorization for the first three products in Europe.
Table 2 | FDA-approved CAR T cell therapies.
| Approved CAR T cell therapy | Brand name | Originally marketed by | Date of approval | Target | Approved for the treatment of |
| Tisagenlecleucel | Kymriah | Novartis | 08/30/2017 | CD19 | B-cell acute lymphoblastic leukemia (ALL); Diffuse large B-cell lymphoma (DLBCL) |
| Axicabtagene ciloleucel | Yescarta | Kite Pharma / Gilead | 10/18/2017 | CD19 | Diffuse large B-cell lymphoma (DLBCL); Follicular lymphoma; Primary mediastinal B-cell lymphoma; High-grade B-cell lymphoma |
| Brexucabtagene autoleucel | Tecartus | Kite Pharma / Gilead | 07/24/2020 | CD19 | Relapsed or refractory Mantle cell lymphoma (MCL); Relapsed or refractory B-cell acute lymphoblastic leukemia (ALL) |
| Idecabtagene vicleucel | Abecma | Bluebird Bio / BMS | 03/26/2021 | BCMA | Relapsed or refractory multiple myeloma |
| Lisocabtagene maraleucel | Breyanzi | Juno Therapeutics / BMS | 02/05/2021 | CD19 | High-grade B-cell lymphoma; Diffuse large B-cell lymphoma (DLBCL); Primary mediastinal B-cell lymphoma; Follicular lymphoma grade 3B |
Application of Live-Cell Imaging in CAR T Cell Research
Role of imaging in CAR T cell research
Although cancer immunotherapy using CAR T cells has made remarkable strides in the past decade, many aspects of this novel therapy remain poorly understood. Further research is required to elucidate why CAR T cell therapy is not very effective against solid tumors or why patients with certain types of B cell lymphomas do not respond to this therapy48. Because CAR T cells act as a “living drug”, real-time monitoring of CAR T cell migration, expansion-kinetics, and cytotoxicity, are essential to assess their therapeutic efficacy in vivo and identify room for further improvement. While the satisfactory clinical response in patients testifies to the effectiveness of a CAR T cell product against a particular medical condition, inadequate responses could be indicative of toxicity development, allowing the treatment to be further tailored, and augment therapeutic benefits. Thus, non-invasive dynamic tracking of CAR T cells is critical to elucidate their interaction with target cells, gain deeper mechanistic insights into the cascade of events leading to tumor regression, and, eventually, translate this therapeutic modality from preclinical models to clinical adoption.
Non-invasive imaging methods to assess CAR T cell therapy
In this section, we will provide a brief overview of the various imaging modalities used for live CAR T cell imaging with reference to the most recent literature published in this area. To date, live CAR T cell imaging has been primarily done using i) optical imaging, ii) radionuclide imaging, and iii) MRI-based imaging methods.
Optical imaging methods
In preclinical cellular imaging, optical techniques predominate other methods for both in vitro and in vivo studies.
In vitro visualization of CAR T cells
Over the past decade, a myriad of in vitro optical imaging techniques have been used to characterize CAR constructs and capture the molecular details underlying CAR immunological synapse formation, signaling, and CAR-mediated cytotoxicity. Among these, confocal microscopy is a relatively simple and one of the most widely used optical imaging modalities for visualizing CARs at high spatial resolution in vitro. In this technique, both effector and target cells are adhered to glass slides and allowed to interact with each other. Thereafter, cells are fixed and visualized using confocal microscopy. Davenport et al. used this approach to demonstrate that the CAR immune synapse (IS) structure is different from the T cell receptor (TCR) synapse, which in turn, facilitated faster killing of target tumor cells49. In another study, Long et al. used confocal microscopy with Cerulean-tagged CAR to demonstrate aggregation of the anti-GD2 CAR on the cell surface50. Later, Xiong et al. used confocal microscopy to evaluate certain characteristics of the CAR-mediated IS, including tumor antigen clustering, lytic granule polarization, and distribution of key signaling molecules within the IS, and thus, predict the effectiveness of CAR T cells in vivo51. Although these studies revealed some key information on the structure and function of CARs, these experiments were mostly done on fixed cells. To obtain more biologically relevant data, a myriad of live-cell imaging technologies have been introduced. These cutting-edge technologies can be used to visualize CARs at the molecular level and evaluate the cancer-killing efficiency of CAR constructs in vitro. Advanced microscopic techniques such as total internal reflection fluorescence microscopy (TIRFM) have been used to characterize the recruitment of CAR microclusters to the CAR IS52. CytoSMART Technologies have also developed novel live-cell imaging microscopes such as CytoSMART Omni and Lux3 BR that can be used to monitor cellular health, viability, colony formation, migration, and cellular responses to external stimuli in real-time. Using these systems, it is possible to gain critical insights into CAR T cells’ morphology, differentiation, proliferation, etc directly inside a cell culture incubator without disturbing the ideal cell culture conditions55.
In vivo visualization of CAR T cells
To visualize CAR T cells at the organismal level, two optical imaging techniques are currently used, namely, bioluminescence imaging (BLI) and two-photon excitation microscopy (TPEM).
Bioluminescence imaging (BLI). This non-invasive optical imaging modality relies on the detection of photons emitted during enzyme (luciferase)-catalyzed oxidation of a molecular substrate (typically, D-luciferin) when the enzyme is expressed in vivo as a molecular reporter54. In many notable studies, BLI has been successfully used to capture the in vivo bio-distribution, trafficking, and expansion kinetics of CAR T cells in a variety of preclinical mouse models56-59. To enable visualization through BLI, CAR T cells have to be co-transduced with luciferase. The luciferase substrate is injected during imaging. Once exposed to the bloodstream, it circulates and diffuses to CAR T cells where it binds to the enzyme luciferase and emits photons48. Although luciferase from the North American firefly (Photinus pyralis; FLuc) is the most common bioluminescent reporter for research purposes60, useful luciferases have also been cloned from other sources such as jellyfish (Aequorea), sea pansy (Renilla; RLuc), corals (Tenilla), click beetle (Pyrophorus plagiophthalamus), and different types of bacteria61. The major advantages of BLI include its high signal-to-noise ratio, high sensitivity and specificity, lack of phototoxicity, ability to simultaneously image multiple subjects, and non-invasive nature. Nevertheless, BLI has limited imaging depth, requires a substrate to work, and the image acquisition is slower than conventional fluorescence imaging.
Two-photon excitation microscopy (TPEM). Two-photon excitation refers to a fluorescence process, which involves the excitation of a fluorophore by simultaneous absorption of multiple (usually two) near-infrared (NIR) photons and emits a single unit of fluorescence. This technique enables three-dimensional imaging of biological specimens in vivo and provides several advantages over conventional confocal microscopy including deeper tissue penetration, superior spatial resolution, and reduced photobleaching62. Among all CAR detection methods at the organismal level, TPEM is the most suitable modality for mechanistic investigation at the cellular level. In a recent study, Cazaux et al. employed intravital two-photon microscopy to track GFP+CD8+ anti-murine CD19 CAR T cells in a syngeneic lymphoma mouse model. Additionally, TPEM was also used to determine calcium flux and detect apoptosis in CAR T cells and cancer cells, respectively, through Förster resonance energy transfer sensors63. In a longitudinal study, Mulazzani et al. combined a model of primary central nervous system lymphoma (PCNSL) with intravital two-photon microscopy to compare GFP+ anti-CD19 CAR T cell infiltration into PCNSL from intravenous and intracerebral CAR T cell injection. Two-photon microscopy revealed that intracerebral injection led to increased CAR T cell infiltration and persistence (up to 159 days), which, in turn, resulted in the eradication of large, established PCNSL, and longer survival64. In several preclinical studies, TPEM has been successfully used to capture the distribution, motility, and functionality of CAR T cells in vivo at the single-cell level while generating novel hypotheses for CAR T cell therapy failure and relapse65. Despite its several advantageous attributes, TPEM cannot be realistically applied for clinical studies and necessitates equipment and infrastructures that might be inaccessible for many labs.
Radionuclide imaging methods
Radionuclide-based imaging including single-photon emission computerized tomography (SPECT) and positron emission tomography (PET) paired with computed tomography (CT) or MRI are widely used for in vivo cell tracking as these techniques offer high sensitivity and a combination of quantitative physiological as well as tomographic information66-68. To enable radionuclide imaging CAR T cells need to be labeled using either a direct or indirect approach (reporter gene strategy). The direct approach involves in vitro labeling of CAR T cells with radionuclides, such as 89Zr-oxine68, 99m-Tc69, or 111In-tropolonate70. The indirect approach entails the expression of an enzyme, receptor or transporter by reporter gene-transduced CAR T-cells71 so that when the probe is intravenously injected for imaging, it accumulates preferentially in CAR T cells. This PET reporter emits positrons, which colocalize with CAR T cells, lose kinetic energy, collide with a nearby electron, annihilate and emit high-energy photons that are eventually captured with a PET scanner. In recent years, several PET reporter/probe pairs have been used to track CAR T cells in vivo72-73. For example, Sellmyer et al. used Escherichia coli dihydrofolate reductase (eDHFR), an enzyme that binds to 18F-labeled trimethoprim (18F-TMP), for imaging anti-GD2 CAR T cells in mouse xenograft models74. PET scans confirmed colocalization between anti-GD2 CAR T cells and GD2+ tumor, which was revalidated with BLI. In addition to cell tracking, PET/CT scans involving the glucose analog fluorine-18 fluorodeoxyglucose (18F-FDG) as a radiotracer has been used for assessing treatment response to CAR T cell therapy in patients with lymphoma75. In a CAR T cell clinical study (NCT0108292676), PET reporter gene/ probe system combining herpes simplex virus type 1 thymidine kinase (HSV1-TK) with 9-[4-[18F] fluoro-3-(hydroxymethyl) butyl] guanine (18F-FHBG) was evaluated on glioma patients. HSV1-TK is a cytosolic viral kinase, which selectively phosphorylates the nucleoside analog 18F-FHBG, an FDA-approved investigational new drug with well-established pharmacology and safety profile in humans. This clinical study involved co-expression of HSV1-TK with an interleukin-13 (IL-13) zetakine CAR in CD8+ T cells to treat seven patients with recurrent high-grade glioma. Blood-brain barrier disruption in glioma allowed 18F-FHBG to diffuse into the tumor. Subsequently, PET scans revealed enhanced signal around the tumor following CAR T cell infusion, which corroborated active trafficking of CAR T cells into the tumor77.
MRI-based imaging methods
MRI is a non-invasive, radiation-free imaging modality that uses magnetic field and strong radio waves to produce detailed images of organs and tissues in the body with high spatial resolution and excellent soft-tissue contrast. Although MRI has lower sensitivity than PET and optical imaging-based techniques, it can quantify a range of physiological, metabolic, and biochemical parameters. To date, most of the studies using MRI for CAR T cell tracking in vivo have used contrast-enhanced techniques, which involves i) T1 agents, that provide positive contrast upon promoting T1-weighted (spin-lattice) relaxation of water78, ii) T2 agents, such as monocrystalline iron oxide nanoparticles (MION) or superparamagnetic iron oxide nanoparticles (SPION) that result in negative signal enhancement79, and iii) ¹⁹F -based contrast agents, such as perfluorinated nanoemulsions80, which enable live-cell tracking via ¹⁹F MRI. With recent advances in multimodal imaging, the exquisite spatial resolution of MRI can be combined with the high sensitivity of PET or optical imaging to possibly provide the most informative assessment of CAR T cell disposition in vivo81. For example, Kiru et al. labeled CAR T cells with an FDA-approved, iron oxide-based nanoformulation (Ferumoxytol) to enable their non-invasive detection and visualization through MRI, photoacoustic imaging (PAT), and magnetic particle imaging (MPI)79.
Conclusion
Numerous CAR T cell therapies are currently under clinical evaluation worldwide. A recent database query (PubMed search) with the keyword “CAR T-cell therapy” returned over 17971 peer-reviewed articles. Out of these, more than 2000 papers were published in the last year itself. Among these, more than 70 studies depicted results from clinical trials. Despite the challenges that the SARS-CoV-2 coronavirus (COVID-19) pandemic has posed on the delivery of cellular therapeutics82, the growing number of publications and trials on CAR T cell-based therapy is a testament to the incredible promise and hope engendered by this new treatment modality. In addition to cancer immunotherapy, some recent preclinical studies of CAR T cells have also demonstrated satisfactory outcomes against autoimmune diseases17. Therefore, a multitude of targets including the non-malignant targets have yet to be researched. Considering the anticipated increase in the use of CAR T cells in the coming years, the radiologists also need to familiarize themselves with the imaging outcomes from these novel therapies, so that they can offer informed and comprehensive guidelines for efficient clinical management.
Related Products
There are currently no products tagged to this resource.